












Docetaxel is an antineoplastic agent belonging to the taxoid family. It is prepared by semisynthesis beginning with a precursor extracted from the renewable needle biomass of yew plants. The Chemical name for Docetaxel is (2R, 3S)-N-Carboxy-3-Phenylisoserine, N-tert-butylester. 13 ester with 5b-20-epoxy-1,2a,4,7b, 13a-hexahydroxytax-11-en-9-one 4-acetate 2- benzoate, trihydrate.
Docetaxel is a white to almost white powder with an empirical formula of C43H53NO1413H2O2 and a molecular weight of 861.9 It is highly lipophilic and practically insoluble in water.
Docetaxel has the following structural formula:

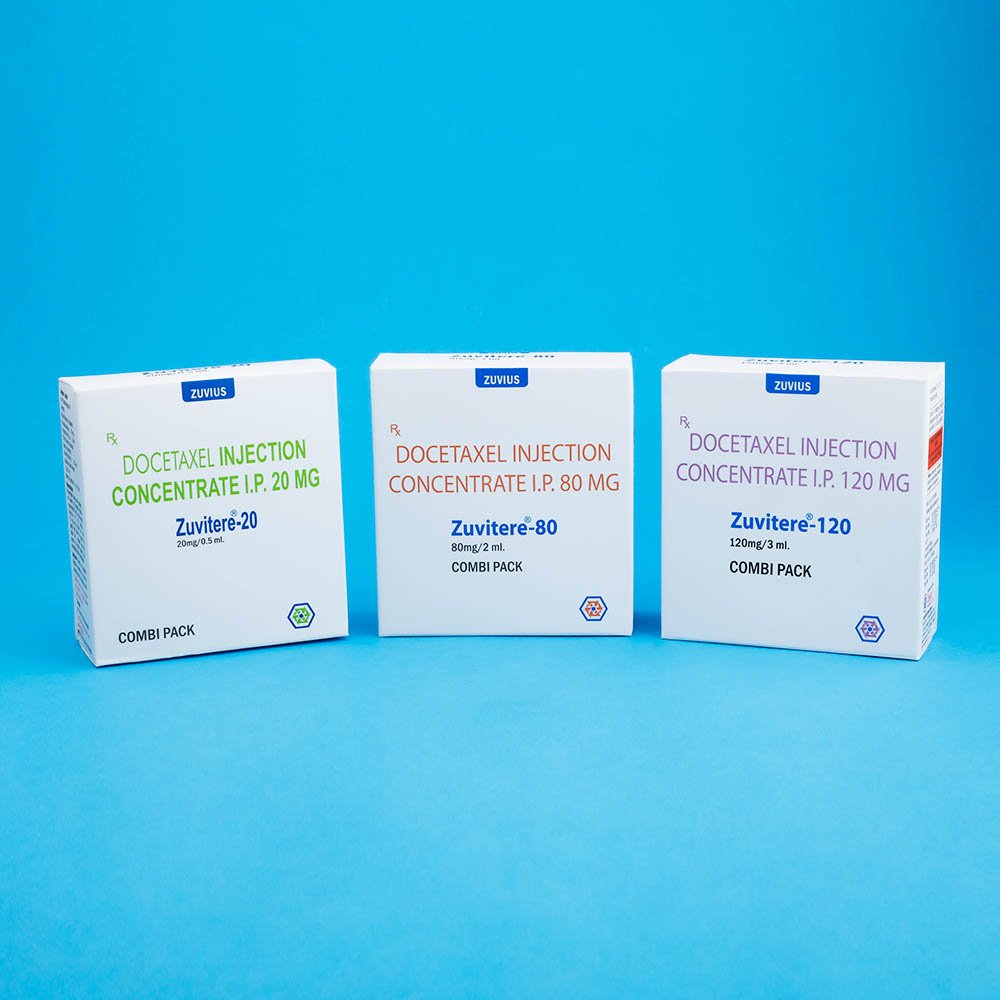
Docetaxel – Non Aqueous Injection 20 mg/0.5 ml, 80 mg/2 ml,120 mg/3 ml.
Composition
Each ml. contains:
Docetaxel Trihydrate I.P.
equivalent to anhydrous Docetaxel 40 mg.
Polysorbate 80 I.P. qs
CLINICAL PHARMACOLOGY
Docetaxel is an antineoplastic agent that acts by disrupting the microtubular network in cells that is essential for mitotic and interphase cellular functions. Docetaxel binds to free tubulin and promotes the assembly of tubulin into stable microtubules while simultaneously inhibiting their disassembly. This leads to the production of microtubule bundles without normal function and to the stabilization of microtubules, which results in the inhibition of mitosis in cells. Docetaxel’s binding to microtubules does not alter the number of protofilaments in the bound microtubbules, a feture, which differs from most spindle poisons currently in clinical use.
The Pharmacokinetics ofdocetaxel has been evaluated in cancer patients after administration of 20-115 mg/m2 in phase 1 studies. The area under the curve (AUC) was dose proportional following doses of 70-115 mg/m2 with infusion times of one to two hours. Docetaxel’s pharmacokinetic profile is consistent with a three compartment pharmacokinetic model, with half-lives for the a, b and g phase of 4 min, 36 min, and 11.1 hr, respectively. This initial rapid decline represents distribution to the peripheral compartments and the late (terminal) phase is due, in part, to relatively slow efflux of docetaxel from the peripheral compartment Mean values for total body clearance and steady state volume of distribution were 21 L/H/m2 and 113 L, respectively. Mean total body clearance for japanese patients dosed at the range of 10-90 mg/m2 was similar to that of European /American populations dosed at 100 mg/m2, suggesting no significant difference in the elimination of docetaxel in the two populations.
A study of “C-docetaxel was conducted in three cancer patients. Docetaxel was eliminated in both the urine and faeces following oxidative metabolism of the tert-butyl ester group, but fecal excretion was the main elimination route within seven days, urinary and fecal excretion accounted for approximately 6% and 75% of the administered radioactivity, respectively. About 80% of the radioactivity recovered in feces is excreted during the first 48 hours as one major and 3 minor metabolites with very small amount (less than 8%) of unchanged drug. Based on in Vitro studies, isoenzymes of the cytochrome P4503A (CYP3A) subfamily appear to be involved in docetaxel metabolism.
A population pharmacokinetic analysis was carried out after docetaxel treatment of 535 patients dosed at 100 mg/m2. Pharmacokinetic prameters estimated by this analysis were very close to those estimated from phase 1 studies. The pharmacokinetics of docetaxel were not influenced by age or gender and docetaxel total body clearance was not modified by pretreatment with dexamethasone. In patients with clinical chemistry date suggestive of mild to moderate liver function impairment (SGOT and / or SGPT) 1.5 times the upper limit of normal (ULN) concomitant with alkaline phosphate >2.5 times (ULN), total body clearance was lowered by an average of 27% resulting in a 38% increase in systemic exposure (AUC)
This average, however, includes a substantial range and there is, at present, no measurement that would allow recommendation for dose adjustment in such patients Patients with combined abnormalities of transaminase and alkaline phosphates should, in general not be treated with ZUVITERE
In Vitro Studies showed that docetaxel is about 94% protein bound, mainly to an aud glycoprotein, albumin, and lipo-proteins. In three cancer patients, the in vitro binding to plasma proteins was found to be approximately 97% Dexamethasone dose not affect the protein binding of docetaxel.
ZUVITERE for Injection concentrate is indicated for the treatment of patients with locally advanced or metastatic breast cancer, who have progressed during anthracycline based therapy or have relapsed
duringanthracycline based adjuvant therapy. It is also indicated for the treatment of advanced or metastatic non-small cell lung cancer affect failure of platinum containing chemotherapy.
For treatment of patients with locally advanced or metastatic carcinoma of the breast after progression during anthracycline-based therapy for metastatic disease or relapse during anthracycline-based adjuvant therapy, the recommended dose of ZUVITERE is 60-100 mg/m2 administered intravenously over one hour every three weeks.
duringanthracycline-based adjuvant therapy, the recommended dose of ZUVITERE is 60-100 mg/m2 administered intravenously over one hour every three weeks.
For treatment of patients with advanced or metastatic non-small cell lung cancer after the failure of prior platinum containing therapy , the recommended dose of docetaxel is 75-100 mg/m2 I.V. over one hour every 3 weeks.
Premedication Regimen : All patients should be premedicated with oral corticosteroids such as
Dexamethasone 16 mg. per day (e.g. 8 mg. BD for 5 days starting 1 day prior to ZUVITERE administration in order to reduce the incidence and severity of fluid retention as well as the severity of hypersensitivity reaction.
Dosage Adjustments During Treatment : Patients who are dosed initially at 100 mg/m2 and who experience either febrile neutropenia neutrophils < 500 cells/mm3, severe or cumulative cutaneous reactions, or severe peripheral neuropathy during therapy should have the dosage adjusted from 100 mg/m2 to 75 mg/m2. If the patient continues to experience these reactions the dosage should either to decreased from 75 mg/m2 to 55 mg/m2 or the treatment should be discontinued, Conversely, patients who are dosed initially at 60 mg/m2 and do not experience febrile neutropenia,neutrophils< 500 /mm2 for more than one week, severe or cummulative cutaneous reactions or severe peripheral neuropathy during docetaxel therapy may tolerate higher doses.
Preparation and Reconstitution of Injection:
Preparation of the Premix solution: Docetaxel for Injection Concentrate require dilution prior to use. A Sterile, non-pyrogenic, single dose diluent containing 13% w/v alchol IP in water for Injection is supplied for that purpose, Remove the appropriate number of vials of docetaxel Injection Concentrate from the refrigerator. Allow the vials to stand at room temperature for approximately 5 minutes. Using a syringe fitted with a needle, aseptically withdraw the entire contents of the solvent for Docetaxel Injections concentrate vial and gently transfer it to the corresponding vial of Docetaxel Injection Concentrate (i.e. 9.9 ml +5% for ZUVITERE – 120 ; 7.33 ml, + 5% for ZUVITERE – 80, 1:83 ml. + 5% ZUVITERE – 20) This will assure a minimal extractable volume of 12 ml. (for ZUVITERE-120) or 8 ml. (for ZUVITERE – 80) or 2 ml. (for ZUVITERE -20) of the premix solution containing 10 mg. Docetaxel / ml. for example, a patient requiring a dose of 140 mg. Docetaxel would require 14 ml. premix solution.
Preparation of the infusion solution: Asceptically withdraw the required amount of ZUVITERE premix solution (10 mg. docetaxel /ml) with a calibrated syringe and inject the required volume of premix solution into a 150 ml. Infusion bag or bottle of either sodium chloride intravenous infusion IP or Dextrose Intravenous infusion IP to produce a final concentration of 0.9 mg/ml.
1) If dose greater than 240 mg. of Docetaxel is required use a large volume of the infusion vehicle so that a concentration of 0.9 mg /ml Docetaxel is not exceeded.
2) Thoroughly mix the infusion by manual rotation.
3) As with all products ZUVITERE should be inspected visually for particulate matter or discoloration prior to administration whenever the solution and container permit. If ZUVITERE for Injection premix solution or infusion solution is not clear or appears to have precipition, the solution should be discarded.
ZUVITERE : Infusion solution should be administered intravenously as a one-hour infusion under ambient room temperature and light conditions contact of the undiluted cncentrate with plasticized PVC equipment or devices used to prepare solutions for infusion is not recommended. In order to minimise patient exposure to the plasticizer DEHP (di-2-ethylhexyl phthalate), which may be leached from PVC infusion bags or sets, diluted ZUVITERE solution should be stored in bottle (glass, polypropylene) or plastic bags (polypropylene, polyolefin) and administered through polyethyle-lined administration.
disease has not been established. )
ZUVITERE (docetaxel) for Injection concentrate should be administer under the supervision of a qualified physician experienced in the use of anti neoplastic agents. Appropriate management of complications is possible only when adequate diagnostic and treatment facilities are readily available.
Toxic Deaths: Docetaxel administered at 100mg/m2 was associated with deaths considered possibly or probably related to treatment in 2.4% (34/1435) of patients with normal liver functions and in 11% (6/55) of Patient with abnormal liver function and is 11% (6/55) of Patient with abnormal liver function (SGOT and / or SGPT>1.5 times ULN together with AP > 2.5 times ULN) Among patients dosed at 60mg.m2, mortality related to treatment occured in 0.6% (3/481) of patients with normal liver function and in 3 to 74 patients with abnormal liver function. Approximately half of these deaths occurred during the first cycle. Sepsis accounted for the majority of the deaths.
Premedication Regimen: Although the optimal premedication regimen is not defined, all patients should be premedicated with oral corticosteroids such as dexamethasone 16mg per day (e.g.), 8 mg BID) for 5 days starting 1 day prior to ZUVITERE to reduce the severity of fluid retentionand hypersensitivity reactions.
Hypersensitivity Reactions: Patients should be observed closely for hypersensitivity reactions, especially during the first and second infusions, severe hypersensitivity reactions characterized by hypotension and /or Bronchospasm, or generalized rashierythemaoccured in 0.9% of patients who received the recommended premedication Hypersensitivity reactions requiring discontinuation of the docetaxel infusion were reported in 5 out of 1260 patients who did not receive premedication. Patients with a history of severe hypersensitivity reactions should not be rechallenged with ZUVITERE
Hematologic Effects: Neutropenia (less than 2000 neutrophils /mm2 occur in nearly all patients given
100 mg/m2, and 75-80% of patients given 60-75 mg/m2 Frequent monitoring of blood counts is, therefore, essential so that dose can be adjusted. ZUVITERE should not be administered to patients with neutrophils <1,500 cell mm2
Three breast cancer patients with severe liver impairment (bilirubin> 1.7 times ULN) developed fatal gastrointestinal bleeding associated with severe drug -induced thrombocytopenia
Hepatic Impairment : ZUVITERE should generally not be given to patients with bilirubin>upper limit of normal (ULN), or to patients with SGOT and /or SGPT> 15X ULN concomitant with alkaline phosphatase 2.5 XULN Patient with elerations of transaminase concurrent with alkaline Phosphatase are at increased risk for the development of grade 4 neutropenia, febrile neutropenia, infections, severe thrombocytopenia, severe stomatitis, severe skin toxicity, and toxic death.
Patient with isolated elevations of transaminase > 1.5 x ULN also had a higher rate of febrile neutropenia grade 4 but did not have an increased incidence of toxic death, Billrubin SGOT or SGPT, and alkaline phosphatase, values should be obtained prior to each cycle of ZUVITERE therapy and reviewed by the treating physician.
Fluid Retention: Severe fluid retention occured in 6% of patients despite use of a 5-day dexamethasone premedication regimen. It was characterized by one or more of the following events. poorly tolerated peripheral edema, generalized edema, pleural effusion requiring urgent drainage, dyspnea at rest, cardiac tamponade, or pronounced abdominal distention (due to ascites) ,
Pregnancy: Docetaxel can cause fetal harm when administered to pregnancy women, Studies in both rats and rabbits at doses equal to or greater than 0.3 and 0.03 mg/kg/day, respectively (about 1/50 and 1/300 the daily maximum recommended human dose on a mg/m2 basis), administered during the period of organogenesis, have shown that docetaxel is embryotoxic and fetotoxic (characterized by intrauterine mortality, increased resorption, reduced fetal weight, and fetal ossification delay). The dose indicated above also caused matemal toxicity. There are no adequate and well-controlled stuides in pregnant women using docetaxel.
If ZUVITERE is used during pregnancy, or if the patient becomes pregnant while receiving this drug, the patient should be apprised of the potential hazard to the fetus or potential risk for loss of the pregnancy. Women of childbearing potential should be advised to avoid becoming pregnant during therapy with ZUVITERE
General: Responding patients may not experience an improvement in performance status on therapy and may experience worsening. The relationship between changes in performance status, response to therapy and treatment – related side effects has been established.
Hemotologic Effects: In order to monitor the occurrence of myelotoxicity, it is recommended that frequent peripheral blood cell counts be performed on all patients receiving ZUVITERE patients should not be retreated with subsequent cycles of ZUVITERE, until neutrophils recover to a level> 1,500 cells.mm3 and platelets recover to a level > 100,000 cells/mm2
A 25% reduction in the dose of docetaxel is recommended during subsequent cycle following severe neutropenia (<500 cells/mm3) lasting 7 days or more, febrile neutropenia, or grade 4 infections in a docetaxel cycle.
Hypersensitivity Reactions: Hypersensitivity reactions may occur within a few minutes following initiation of an ZUVITERE infusion. If minor reaction such as flushing or localized skin reactions occur, of ZUVITERE and aggressive therapy. All patients shouldbepremedicated with an oral corticosteroid prior to the initiation of the infusion of ZUVITERE
Cutaneous: Localized erythoma of the extremities with edema followed by desquamation has been observed. In case of severe skin toxicity, an adjustment in dosage is recommended (See Dosage and Administration section) Fluid REtention: Severe fluid retenation has been reported following docetaxel therapy. Patients should be premedicated with oral corticosteroids prior to each administration to reduce the incidence and severity of fluid retention (see Dosage and Administration section). Patients with pre-existing effusions should be closedly monitored from the first dose for the possible exacerbation of the effusions. In patients who received the recommended premedication, moderate fluid retention was completely, but sometimes slowly reversible following discontinuation of docetaxel (median of 29 weeks). The median cumulative dose to onset of moderate or severe fluid retention was 7052 mg/m2 in patients receiving premedication. Patients developing peripheral edema may be treated with standard measure, e.g., salt restriction, oral diuretic (S)
Neurologic : Severe neurosensory symptoms (paresthesia, dysesthesia, pain) were observed among 7% of 134 patients with anthracycline-resistant breast cancer. When these occur, dosage must be adjusted. if symptoms persist, treatment should be discontinued (see Dosage Administration section). Patients who experienced neurotoxicity in clinical trials and for whom follow-up information on the complete resolution of the event was available has spontaneous reversal of symptoms within a median of 9 weeks from onset (range : 0 to 106 weeks) and only about 3.8% of patients required discontinuation due to neurotoxicity. Peripheral motor neuropathy mainly manifested as distal extremity weakness occured in 13.4% ( 7.1% severe) of the 127 anthracycline-resistnat breast cancer patients with normal LFTs. No neuromotor toxicity was reported in the 7 patients with elevated LFTs.
Extremity weakness occured in 13.4% (7.1% severe) of the 127 anthracycline-resistant breast cancer patients with normal LFTs. No neuromotor toxicity was reported in the 7 patients with elevated LFTs.
Asthenia: Severe asthenia has been reported in 111% of the patients but has led to treatment discontinuation in only 2.6% of the patients. Severe asthenia was reported in 23% of 134 patients with anthracycline-resistant breast cancer and 5.5% of the 786 cycles received. Symptoms of fatigue and weakness may last a few days up to several weeks and may be associated with deterioration of performance status in patients with progressive disease.
There were 1495 patients enrolled in 37 clinical trials conducted in North American and Europe (624 breast carcinoma patients and 866 patients with other tumor types) who received docetaxel at an initial dose of 100 mg/m2 every 3 weeks. Five patients were not evaluable for toxicity since they discontinued docetaxel treatment due to acutehypersensitivity reactions with the first infusion. At least 95% of these patients did not receive hematopoietic support.
Hematologic: Bone marrow suppression is the major dose-limiting toxicity of docetaxel. Neutropenia is reversible and not cumulative. The median day to nadir was 8 days, while the median duration of severe neutropenia <500 cells mm 2) was 7 days. Among patients with normal liver function treated with docetaxel, severe neutropenia occured in 76% and lasted for more than 7 days in 4.3% of cycles, anemia was reported in 89.5% with severe cases being reported in 8.4% of the patients)
Febrile neutropenia (<500 cells/mm2 with IV antibiotics and / or hospitalization) occured in 11.8% of
the patients with normal liver function (3% of the cycles) Infections episodes occured in 21.7% of the patients (6.2% of the cycles) and were fatal in 1.6% of those treated with docetaxel (1.4% in breast cancer patients) Thrombocytopenia (<100,000 cells/mm2) occured in 7.5% of the patients with normal liver function. Bleeding episodes were reported in 2.3% of the patients. A fatal gastrointestinal hemorrhage associated with thrombocytopenia was reported in one patients. Three breast cancer patients with severe liver impairment (billrubin>1.7 times ULN) developed fatal gastrointestinal bleeding associated with severe drug induced thrombocytopenia.
Hypersensitivity Reactions: Hypersensitivity reactions requiring discontinuation of the docetaxel infusion were reported in 5 patients of 1260 who did not receive premediction. Severe hypersensitivity reactions characterized by hypotension and / or bronchospasm, or generalized rash/erythemas have been observed in only 0.9% of patients with normal liver function receiving the recommended premedication regimen and none of these patients had to discontinue therapy.
Minor events, including flushing, rash with or without pruritis, chest lightness, back pain, dyspnes, drug fever, or chills, have been reported and resolved afer discontinuing the infusion and appropriate therapy.
Fluid Retention: Events such as edema and weight gain and, less frequently, pleural effusion, pericardial affusion or asscites have been described. Among 229 patients with normal liver function receiving the recommended pretreatment, severe fluid retention was observed in 6% causing treatment discontinuation in 1.7% when it occurs, peripheral adema usually starts at the lower extremities and may become generalized with a median weight gain of 2 kg. Fluid retention is cumulative in incidence and severity. The median cumulative dose to onset of moderate or severe fluid retention was 705 mg/m2 . Fluid retention was completely reversible, resolving a median of 29 weeks (range : 0 to 42 + weeks) from the last docetaxel infusion.
Cutaneous: Reversible cutaneous reactions characterized by a rash including localized eruptions, mainly on the feet and / or hands, but also on the arms, face or throat, usually associated with pruritus, have been observed. Eruptions generally occurred within one week after docetaxel Infusion, recovered before the next infusion and were not disabling. Severe symptoms, such as eruptions followed by desquamation, occurred in 5.6% of the patients and rarely led to interruption or discontinuation of docetaxel treatment. Alopacia occurred in 80% of patients, and it was severe in 61.8% of patients.
Severe nail disorders occurred in 2.6% of the patients. These reactions were characterized by hypo-orhyperpigmentaion, and occasionally by onycholysis (in 0.8% of patients) and pain.
Neurologic : Neurosensory symptoms characterized by paresthesia, dysesthesia or pain (including burning sensation) have been reported in patients receiving docetaxel. Severe reactions were observed in 3.9% Neuromotor events characterized mainly by weakness have been reported and were severe in 3.7% of the patients.
Gastrointestinal : Gastrointestinal reactions (nausea, and / or vomiting, and / or diarhoea) were generally mild to moderate and severe reactions occured in 8.2% of the patients. Stomatitis was reported in 42.3 & of patients receiving docetaxel. Severe reactions were observed in 5.3% of patients.
Cardiovascular: Hypertension occured in 3.6% of the patients, 3.4% required treatment. Clinically meaningful events such as heart failure, sinus tachycardia, atrial, flutter, dysrhythmia, unstable angina, pulmonary edema, and hypertension occured rarely.
Infusion Site Reaction: Infusion site reactions were generally mild and considered of Hyperpigmentation, inflammation, redness or dryness of the skin, phlebitis, extravesation, or swelling of the vein.
Hepatic: In patients with normal LFT’s at baseline, bilirubin values greater than the ULN occured in 8.9% of patients, increase in SGOT or SGPT> 1.5 times the ULN, or alkaline phosphatase> 2.5 times ULN, were observed in 18.1% and 7.6% of patients, respectively. During the study, increases in SGOT and / or SGPT> 1.5 times ULN concomitant with alkaline phosphatase > 2.5 times ULN occured in 4.5% of patients with normal LFT’s at baseline, (Whether these changes were related to the drug or underlying
There have been no formal clinical studies to evaluate the drug interactions of docetaxel with other medications. In vitro studies have shown that the metabolism of docetaxel may be modified by the concomitant administration of compounds that induce, inhibit or are metabolized by cytochrome P450 3A4, such as cyclosporine, terfenadine, ketoconazole, erythromycin, and troleandomycin, Caution should be exercised with these drug when treating patients receiving as there is a potential for a significant interaction.
CARCINOGENICITY, MUTAGENICITY, IMPAIRMENT OF FERTILITY: No studies have been conducted to assess the carcinogenic potential of docetaxel. Docetaxel has been shown to be clastogenic in the vitro chromosome aberration, test in CHO-K1 cells and in the in vivo micronucleus test in the mouse, but it did not induce mutagenicity in the Ames test, or the CHO/HGPRT gene mutation assays. Docetaxel produced no impairment of fertility in rats when administered in multiple I.V. dose of up to 3mg./kg (about 1/50 the recommended human dose on a mg/m2 basis), but decreased testicular weight were reported.
This correlates with findings of a 10-cycle toxicity study (during once every 21-days for 6 months) in rats and dogs in which testicular atrophy or degeneration was observed at I.V. doses of 5 mg/kg in rats
and 0.375 mg/kg in dogs (about 1/3 and 1/15 the recommended human dose on a mg/m3 basis, respectively). An increased frequency of dosing in rats produced similar effects at lower dose levels.
Nursing Mothers: It is not known whether ZUVITERE is excreted inhuman milk, and because of the
potential for serious adverse reactions in nursing infants from ZUVITERE, mothers should discontinue nursing prior to taking the drug.
Padiatric use: The safety and effectiveness of ZUVITERE in pediatric patients have been established.
There is no known antidote for docetaxeloverdosage. In case of overdosage, the patient should be kept in a specialized until where vital functions can be closely monitored. Anticipated complications of overdosage include: bone marrow suppression, peripheral neurotoxicity, and mucositis.
There are two reports of overdose. One patients received 150mg/m2 and the other received 200 mg/m2 as one hour infusions. Both patients experienced severe neutropenia, mild asthenia, cutaneous reactions, and mild paresthesia and recovered without incident. In mice, lethality was observed following single I.V. : neurotoxicity associated with Paralysisnoneytrision of hind limbsand myelin digeneration was observed in mice at 48 mg/kg (about 1.5 times the
srommended human do on a mg/m2 basis) doses that were > 154 mg /kg (about 4.5 times the recommended human dose on a mg/m2 basis) In male and female rats, lethality was observed at a dose of 20 mg/kg . (Comparable to the recommended human dose on a mg/m2 basis ) and was associated with abnormal mitosis and necrosis of multiple organs.
CONTRAINDICATIONS :
ZUVITERE is contraindicated in patients who have a history of severe hypersensitivity reactions to docetaxel or to other drugs formulated with polysorbate 80. ZUVITERE should not be used in patients with neutrophil counts < 1,500 cells/mm2
The unopened vials should be stored under refrigeration at a temperature of 2-8°C (36-46°F) and should be protected from light. Freezing dose not adversely affect the product.
Solvent for Docetaxel Injection Concentrate should be stored below 25°C. protected from light. ZUVITERE premix solution (10 mg Docetaxel/ ml. ) and fully prepared ZUVITERE infusion solution (in either sodium chloride intravenous infusion IP or Dextrose intravenous IP ) should be used as soon as possible after preparation . However, the premix solution is stable for 8 hours either at room temperature, 15° to 25° C (59°to 77°F), or stored refrigerated, 2° to 8°c (36° to 46°F)
WARNING:
To be sold by retail on the prescription of a cancer specialist/cancer Hospital and institution only.
Keep out of reach of children.
HANDLING:
ZUVITERE is a cytotoxic anticancer drug and as with other potentially toxic compounds, caution should be exercised when handling and preparing ZUVITERE solutions. The use of gloves is recommended. If ZUVITERE concentrate, premix solution, or infusion come into contact with the skin or mucosa immediately and throughly wash with soap and water.
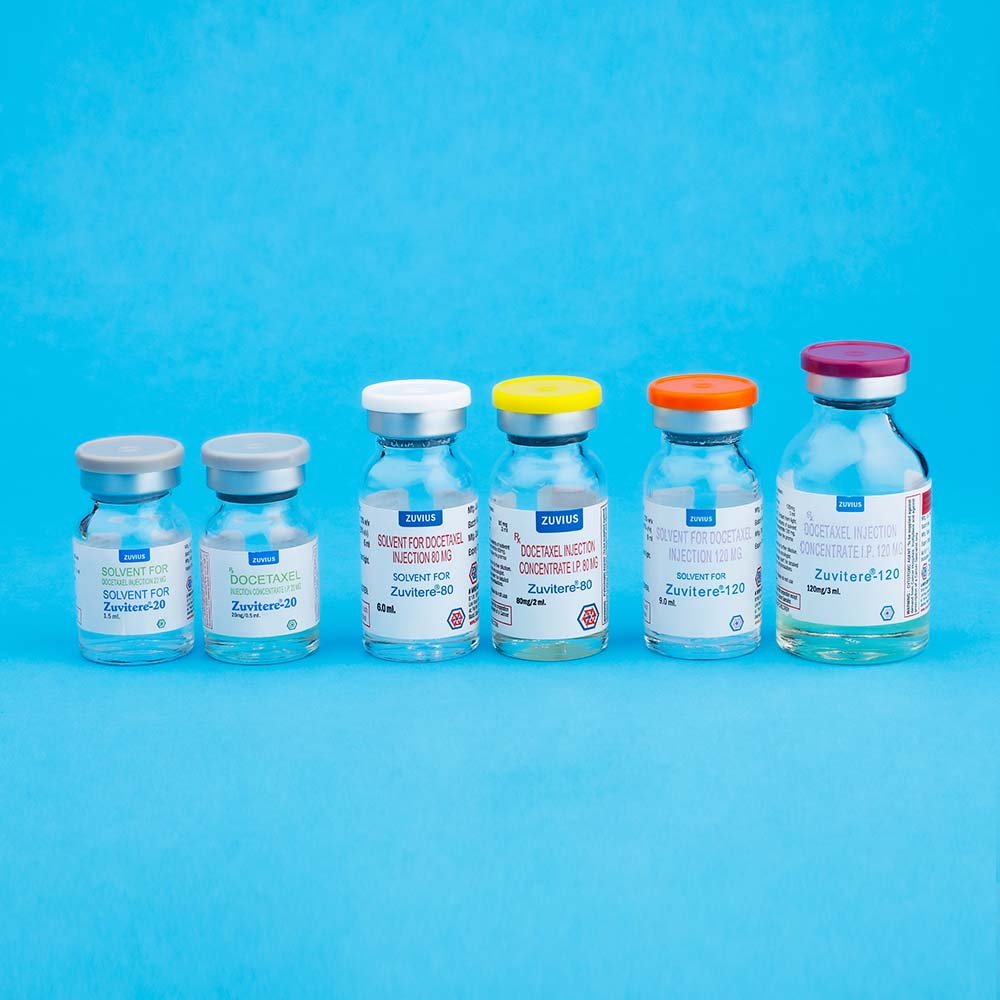
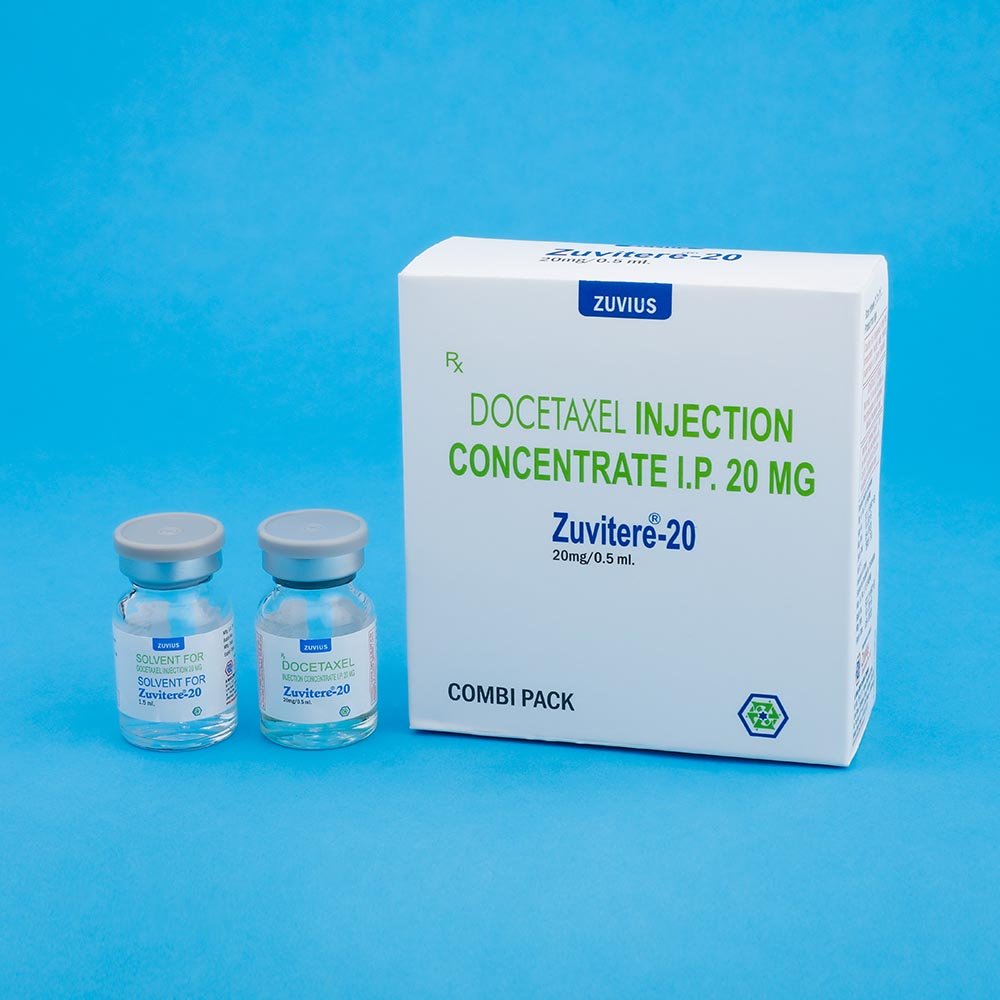
ZUVITERE-20 :
Combipack contains:
1) One vial of Docetaxel Injection Concentrate (20 mg/0.5 ml) with insert
2) One vial of Solvent for Docetaxel Injection Concentrate (1.5 ml) with insert
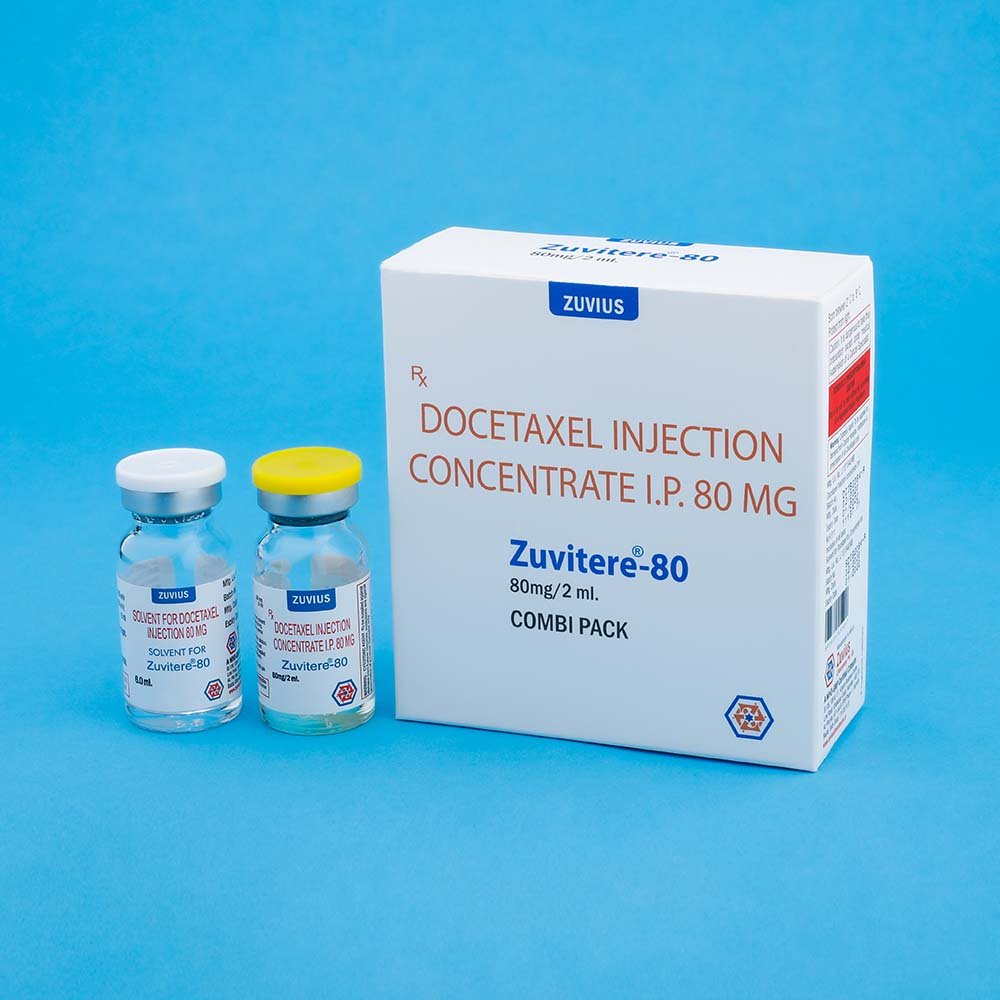
ZUVITERE-80 :
Combipack contains:
1) One vial of Docetaxel Injection Concentrate (80 mg./2.0 ml.) with insert
2) One vial of Solvent for Docetaxel Injection Concentrate (6.0 ml) with insert
ZUVITERE-120 :
Combipack contains:
1) One vial of Docetaxel Injection Concentrate (120 mg/3.0 ml) with insert
2) One vial of Solvent for Docetaxel Injection Concentrate (9.0ml) with insert








