











Zetabin - Gemcitabine Inj
Gemcitabine Inj
Strength: 1GM, 1.4GM, 200MG
Pack Size: 1 vial
Drug Class: Anthracyclines and related substances
Dosage and Administration:
Pancreatic Adenocarcinoma
Adults:
I.V. 1,000 mg/m2once weekly for 7 wk followed by 1 wk of rest.
Dosage Adjustment:
Based on degree of hematologic toxicity. If marrow suppression is detected, modify gemcitabine dosage as follows; absolute granulocyte count (AGC) is at least 1,000 X 106 /L and platelet count is at least 100,000 x 106/L give 100% of full dose; agc is 500 to 999 x 106 /L or platelet count is 50,000 to 99,999 x 106 /L, give75% of full dose; AGC is less than 500×106 /L or platelet count is less than 50,000 x106 /L, hold dose.
Subsequent cycles.
Give the same dose once weekly for 3 consecutive wk followed by 1 wk of rest. After at least 7 doses ( 1 cycle), the dose may be increased to 1,250 mg/m2 once weekly for 3 wk, followed by 1 wk of rest, if the following criteria are met.
– Nonhematologic toxicity is no greater than WHO grade 1,
– platelet nadirs are greater than 100,000×106 /L
– AGC nadir is more than 1,500 x 106 /L
If patient still meets the above creteria after receiving 3 doses of the higher regimen, gemcitabine dose may be increased to 1,500 mg/m2 I.V. once weekly for 3 wk, followed by 1 wk of rest.
NSCLC
Adults
I.V. In combination with cisplatin, gemcitabine 1,000 mg/m2 on days 1,8 and 15 of each 28-day cycle Alternatively, gemcitabine 1,250 mg/m2 may be given
I.V. on days 1 and 8 of a 21 day cycle.
Dosage adjustment
Reduce or delay the gemcitabine dose in patients with neutropenia or thrombocytopenia on the day of treatment. On the day of the Schedule dose, if AGC is 500 to 999 x106 /L or platelet count is 50,000 to 99,999 x 106 /L, give 75 % of the prior dose. If AGC is less than 500 x 106 /L or platelet count is less than 50,000 x 106 / L, hold dose. For grade 3 or 4 nonhematologic toxicity, hold gemcitabine or reduce the dose 50% in patients with NSCLC. Dosage reduction is not required for severe alopecia or nausea and vomiting. Dosage reduction may be necessary in renal or hepatic function impairment. Use additional caution in these patients.
Ovarian Cancer Adults
I.V. 1,000 mg/m2 over 30 min on days 1 and 8 of each 21- day cycle in combination with carboplatin. Monitor prior to each dose with a CBC, including differential counts. Prior to each cycle, the AGC should be least 1,500 x 106/L and the platelet count should be at least 100,000x 106 /L
Dosage Adjustment
Based on granulocyte and platelet counts on day 8 of therapy. If marrow suppression is detected, modify gemcitabine dosage as follows: AGC is 1,000 to 1,499×106/L and/or platelet count is less than 75000 to 99,999 x106/L give 50% of full dose, AGC is less than 1000 x 106 /L and /or platelet count is less than 75,000 x 106 /L, hold dose. For severe nonhematologic toxicity, except nausea/ vomiting, hold gemcitabine therapy or decrease 50%.
Subsequent cycles.
Dose adjustments for gemcitabine in combination with carboplatin are based upon observed toxicity. Reduce the gemcitabine dose to 800 mg/m2 on days 1 and 8 if-any of the following hematologic toxicities are present:
-AGC less than 500×106 /L for more than 5 days,
– AGC less than 100×106 /L for more than 3 days,
-Febrile neutropenia,
– Platelet count less than 25,000 x 106 /L
– Cycle delay of more than 1 wk because of toxicity.
If any of the above recur after the initial dose reduction, for the subsequent cycle, give gemcitabine 800 mg/m2 on day 1 only.
Breast Cancer
Adults
I.V. 1,250 mg/m2 over 30 min on days 1 and 8 of each 21-day cycle in combination with paclitaxel. Monitor prior to each dose with a CBC, including differential . Prior to each cycle, AGC should be at least 1,500 x 106/L and the platelet count should be at least 100,000 x 106/L.
Dosage adjustment
Based on granulocyte and platelet counts on day 8 of therapy. If marrow suppression is detected, modify gemcitabine dosage as follows: AGC is 1,000 to 1,199 x 106/L or platelet count is 50,000 to 75,000 x 106/L , give 75% of full dose; AGC is 700 to 999 x 106/L and platelet count is less than 50,000 x 106 /L, give 50% of full dose, AGC is less than 700x 106/L hold gemcitabine. For severe nonhematologic toxicity, except alopecia and nausea / vomiting, hold gemcitabine therapy or decrease 50%.
General Advice
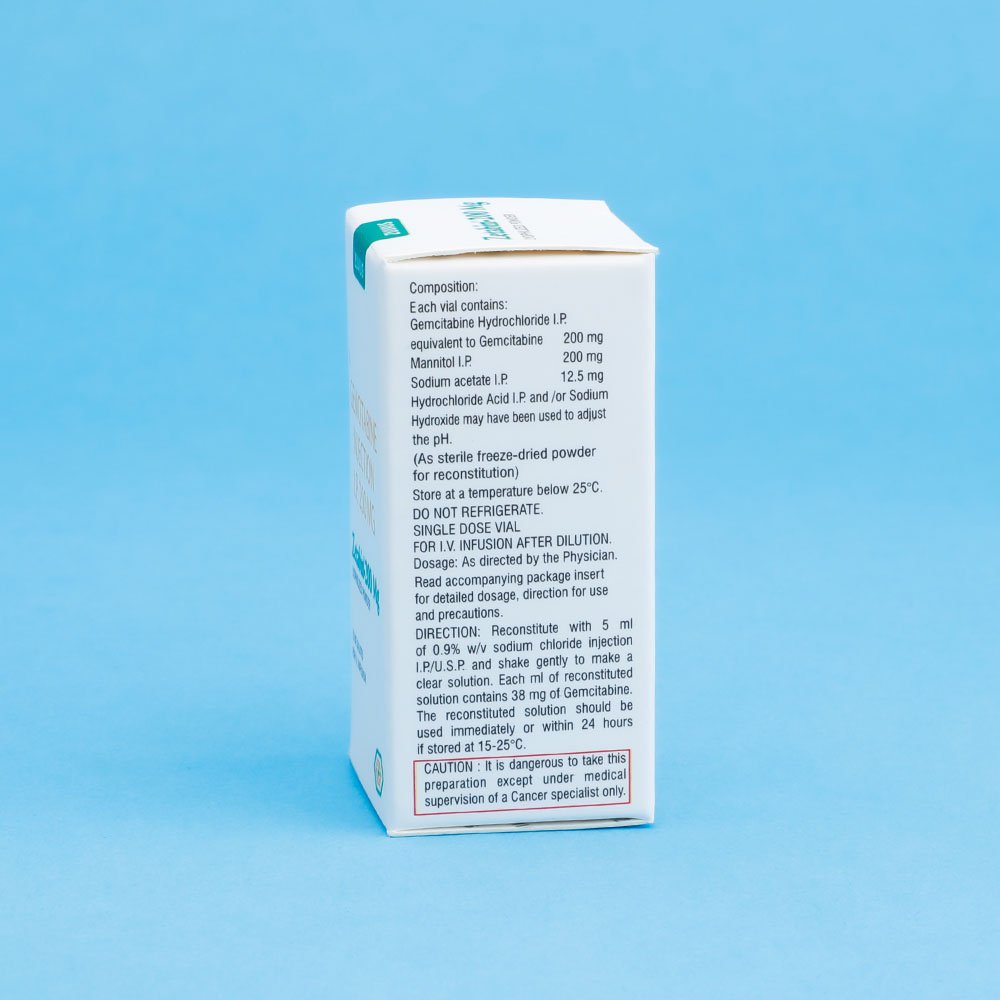
– Reconstitute with preservative-free sodium chloride 0.9% .
– Reconstituted solution may be administered directly or further diluted with sodium chloride 0.9% to a final gemcitabine concentration at least 0.1 mg/ml.
– Administer by I.V. infusion over 30 min. Prolonging infusion past 60 min increases the risk of myelosuppression.
– Reconstituted gemcitabine is a clear, colorless to light straw-colored solution. Do not administer if particulate matter or discoloration is found.
Cold Storage: no
Gemcitabine HCL is a nucleoside analogue that exhibits antitumor activity.
Gemcitabine HCL is 2-deoxy-2,2- difluorocytidinemonohydrochloride (Beta-isomer).
The Structural Formula is as follows:

The empirical formula for Gemcitabine HCL is C9H11F2N3O4. HCl. It has a molecular weight of 299.66.
Gemcitabine HCl is a white to off-white solid. It is soluble in water, slightly soluble in menthanol, and practically insoluble in ethanol and polar organic solvents.
First-line treatment of locally advanced or metastatic Pancreatic adenocarcinoma in patients previously treated with 5-fluorouracil; first-line treatment of locally advanced or metastatic non-small cell lung cancer (NSCLC);
Treatment of advanced ovarian cancer that has relapsed at least 6 mo after completion of platinum- based therapy; first-line treatment of metastatic breast cancer after failure of prior anthracycline-containing adjuvant chemotherapy, unless anthracyclines are clinically contraindicated.
Unlabeled Uses
Treatment of biliary cancer, bladder cancer, relapsed or refractory testicular cancer, squamous cell carcinoma of the head and neck.
Usage:
Reconstitute with Sodium Chloride Injection I.P. and shake gently to make a clear solution.
Gemcitabine kills cells undergoing DNA synthesis and blocks the progression of cells through the G1/S-phase boundary. Gemcitabine is metabolized by nucleoside kinases to diphosphate (dFdCDP) and triphosphate (dFdCTP) nucleosides. It works by slowing or stopping the growth of cancer cells in your body.
- Read the label carefully before use
- Do not exceed the recommended dose
- Monitor Prior to each dose, monitor with a CBC, including differential and platelet count; perform laboratory evaluation of renal and hepatic function (including serum creatinine, potassium , calcium, and magnesium) prior to starting therapy and periodically there after. Pregnancy Category D Lactation Undetermined Children Safety and efficacy not established. Renal Function Hemolytic uremic syndrome and renal failure have been reported. Renal failure leading to death or requiring dialysis, despite discontinuing therapy, has been reported. Use with caution in patients with preexisting renal function impairment. Hepatic Function Serious hepatotoxicity, including liver failure and death, has been reported. Use with caution in patients with preexisting hepatic function impairment. Hematology Myelosuppression, Including anemia, leukopenia, and thrombocytopenia, is usually the dose-limiting adverse reaction for therapy. PulmonaryPulmonary toxicity has been reported. In case of severe toxicity, discontinue therapy Immediately and institute supportive measures.




